22 fev Diagnóstico da Fibrose Pulmonar Idiopática (FPI)
Tiempo de lectura: 10 minutosÀ semelhança de outras doenças pulmonares, as manifestações usuais da FPI incluem a presença de tosse e dispneia progressiva aos esforços. Como a doença afeta pacientes com idade mais avançada, na maioria das vezes acima de 60 anos de idade, a falta de ar é atribuída aos efeitos do envelhecimento ou ao sedentarismo, retardando o diagnóstico.
O tempo de história varia em geral de 6 a 24 meses até́ o diagnóstico. Ocasionalmente, a doença é detectada na fase assintomática.
A dispneia deve ser caracterizada, no momento da avaliação inicial, por escalas padronizadas, como a da escala modificada do Medical Research Council (mMRC) (tabela 2), desde que sua intensidade é fator prognóstico independente da função pulmonar e grau de hipoxemia, além de ter
correlação estreita com a qualidade de vida.4, 30 Ansiedade e depressão são também frequentes.

O exame físico demonstra estertores em velcro nas bases pulmonares em 90% dos casos, incluindo a doença em fases mais iniciais. A presença dos estertores em velcro é um achado quase universal, porém não é específico. No entanto, pode ser importante para detectar a doença antes do aparecimento de faveolamento. A presença de hipocratismo digital indica doença avançada, e é hoje observada em menos de 20% dos casos. Achado de hipertensão pulmonar como hiperfonese de B2 em foco pulmonar, turgência jugular e edema em membros inferiores podem ser observados em fases tardias da doença.
A FPI ocorre principalmente em adultos mais velhos, é limitada aos pulmões e está associada ao padrão histopatológico e/ou radiológico de pneumonia intersticial usual (PIU). A presença de estertores em velcro é indicação para realização de TCAR, sem contraste, incluindo cortes em
decúbito ventral para detecção de doença precoce. Uma apresentação clínica compatível e o padrão característico na TCAR, incluindo a presença de faveolamento, são sufi cientes para o diagnóstico ser considerado definitivo, sem necessidade de realizar biópsia pulmonar cirúrgica (BPC). Porém, mesmo com os achados típicos na TCAR, é muito importante lembrar que o diagnóstico de FPI requer a exclusão de todas as outras causas conhecidas de DPIs que possam resultar em padrão PIU, incluindo pneumonite de hipersensibilidade (PH), colagenoses (mesmo sem achados clínicos aparentes) e lesão pulmonar por drogas.3
Para o diagnóstico de FPI, a sensibilidade da TCAR é maior do que seu valor preditivo positivo, isto é, nem todos os pacientes com FPI terão TCAR típica, porém quando os achados típicos estão presentes, preenchidas as condições acima, o diagnóstico clínico é aceito.1
Histologicamente, o padrão de PIU é caracterizado pela presença de fibrose de distribuição heterogênea, em regiões subpleural e parasseptal, associada à “fibrose jovem”, caracterizada pela presença de focos fibroblásticos, e “fibrose avançada”, com deposição de colágeno do tipo I.3
A TCAR na PIU, conforme a sugestão da American Thoracic Society, da European Respiratory Society, da Japanese Respiratory Society, e da Latin American Thoracic Society (ATS/ERS/JRS/ALAT) de 20183, é mostrada na tabela 3 a seguir:

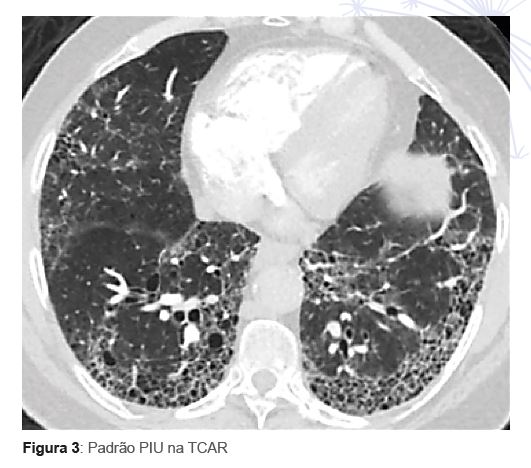
A fi gura 3 ilustra uma TCAR com padrão PIU típico (com presença de faveolamento). O padrão de PIU provável foi sugerido como sufi ciente para o diagnóstico, novamente no contexto clínico apropriado (idosos, em geral ex-fumantes, sem outras causas aparentes para DPI). Difere do padrão
típico pela ausência de faveolamento.
Entretanto, no Brasil, onde a PH é frequente, este padrão isoladamente não deve ser considerado suficiente para o diagnóstico de FPI e BPC (Biópsia Pulmonar Cirúrgica) deve ser considerada para diagnóstico. O padrão chamado de indeterminado envolve duas apresentações: um padrão
reticular discreto, sem bronquiectasias de tração (“padrão incipiente”) e um segundo padrão, mais extenso, mas não classifi cável (“indeterminado verdadeiro”).
Finalmente, o padrão denominado de “diagnóstico alternativo” envolve diversos achados que, quando presentes, devem levar à suspeita de outras condições, embora possa eventualmente ser encontrado como expressão da PIU.
TCAR – Tomografi a computadorizada de alta resolução; PIU – Pneumonia Intersticial Usual; DPI – Doenças Pulmonares Intersticiais; FPI – Fibrose Pulmonar Idiopática.
Quando material de biópsia for considerado necessário para diagnóstico, diversas considerações devem ser feitas. Geralmente isso acontece quando os achados tomográficos não são característicos (o que ocorre em 20 a 30% dos casos) e outra condição é possível pelos dados clínicos (ex: exposição compatível com PH). Também em pacientes com menos de 50 anos, independente dos achados tomográfi cos, a BPC se faz necessária para comprovação diagnóstica.
A broncoscopia com biópsia transbrônquica (BTB) e lavado broncoalveolar (LBA) tem um papel limitado na FPI. Achados sugestivos de FPI na BTB (incluindo focos fi broblásticos e fi brose heterogênea) podem ser observados ocasionalmente, mas não justificam a realização de broncoscopia
de rotina. O LBA pode revelar aumento de neutrófi los (>3%) e/ou eosinófi los, mas doenças pulmonares fibrosantes em geral podem exibir este padrão.1
Mais recentemente foi desenvolvida uma técnica para obtenção de fragmentos histológicos maiores, através do broncofi broscópio, denominada criobiópsia transbrônquica. Nesta, a extremidade distal de uma sonda especial, introduzida através do canal do broncoscópio, é rapidamente
congelada pela liberação de um gás em alta pressão, com resultante aderência ao tecido pulmonar. A retirada da sonda com o broncoscópio permite a retirada de fragmentos maiores e sem artefatos de esmagamento. Estudo comparativo, nos mesmos pacientes, da criobiópsia com BPC obtida por
videotoracoscopia, mostrou que o grau de concordância é elevado.31 Em casos eventuais, entretanto, pela disposição periférica e subpleural da PIU, o diagnóstico não é fechado.
BPC por videotoracoscopia permite a retirada de fragmento grandes de lobos diversos. Quando considerada para diagnóstico em doenças intersticiais fibrosantes, diversos fatores devem ser levados em conta. Idade avançada (acima de 75-80 anos), disfunção pulmonar importante (CVF<
50%, DCO < 35%, SpO2 de repouso < 89%), presença de hipertensão pulmonar e comorbidades diversas são contraindicações.1 A mortalidade pós-operatória, em BPC por videotoracoscopia eletiva, em DPIs, situa-se em torno de 1%.
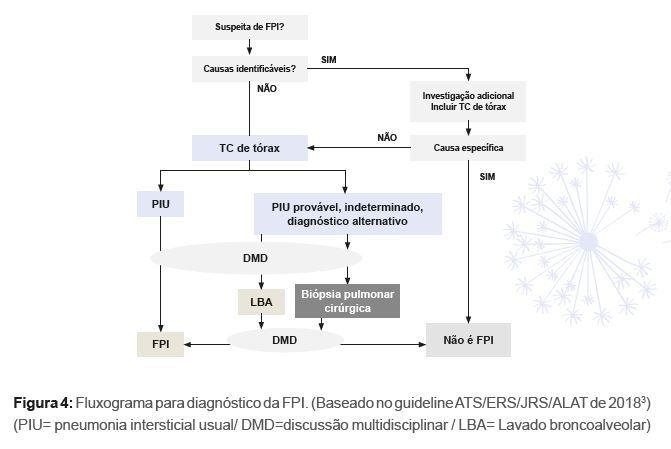
Por fim, recomenda-se que uma discussão multidisciplinar, envolvendo pneumologistas ou clínicos, radiologistas e patologistas, seja feita, tanto na avaliação inicial como após a eventual obtenção de material de BPC para defi nição diagnóstica de FPI.3 Um fl uxograma para diagnóstico de
FPI é apresentado na figura 4.
Impactos da Doença
A FPI é uma doença crônica e progressiva com um impacto signifi cativo na qualidade de vida (QV) do paciente. À medida que a doença progride, ocorre piora progressiva da dispneia associada com declínio importante da função pulmonar, resultando ao fi nal em incapacidade para atividades
habituais e necessidade crescente do uso de oxigênio. Os principais problemas relacionados a QV destes pacientes, portanto, costumam ser: fadiga, prejuízo da mobilidade, dificuldade de realização das atividades de vida diária, perda da capacidade de trabalho, piora da tosse e dispneia acentuada.
A exaustão física anormal e a fadiga são frequentemente consideradas uma consequência natural das doenças pulmonares crônicas. A combinação de mau prognóstico, incerteza no curso da doença e carga acentuada de sintomas afeta fortemente a QV, tanto para pacientes quanto para os membros da família.32
Em pacientes com FPI, as comorbidades, tais como doenças cardiovasculares e tromboembólicas, depressão, distúrbios do sono, câncer de pulmão e diabetes, são frequentes e podem contribuir de maneira importante para a carga de sintomas e pior impacto na QV. A identifi cação e o tratamento de comorbidades podem melhorar a QV e potencialmente infl uenciar o prognóstico.33
Recentes avaliações de QV e as novas modalidades terapêuticas mudaram o manejo da doença, de tal forma que aumentaram a necessidade de avaliações socioeconômica na FPI. O alto custo relacionado à doença inicia-se antes do diagnóstico, pela sua alta prevalência de diagnóstico
tardio e/ou tratamentos baseados na suspeita de outras doenças pulmonares, como infecção de repetição e doença pulmonar obstrutiva crônica (DPOC). São comprovadas as elevadas taxas de exacerbações agudas com necessidade de hospitalizações, além de outras despesas relacionadas
à saúde dos pacientes portadores de FPI como oxigenoterapia e cuidados paliativos na fase mais avançada. Portanto, estratégias e terapias de gerenciamento aprimoradas devem ser planejadas para reduzir custos relacionados à saúde com melhorias nos cuidados de suporte e avaliações
socioeconômica do custo e benefício do tratamento antifibrótico.34, 35
Referências
1. Baldi BG, Pereira CA, Rubin AS, Santana AN, Costa AN, Carvalho CR, et al. Highlights of the Brazilian Thoracic Association guidelines for interstitial lung diseases. J Bras Pneumol. 2012;38(3):282-91.
2. Baddini-Martinez J, Ferreira J, Tanni S, Alves LR, Cabral Junior BF, Carvalho CRR, et al. Brazilian guidelines for the pharmacological treatment of idiopathic pulmonary fibrosis. Official document of the Brazilian Thoracic Association based on the GRADE methodology. J Bras Pneumol. 2020;46(2):e20190423.
3. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198(5):e44-e68.
4. Soares MR, Pereira C, Ferreira R, Nei Aparecida Martins Coletta E, Silva Lima M, Muller Storrer K. A score for estimating survival in idiopathic pulmonary fibrosis with rest SpO2>88. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32(2):121-8.
5. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788-824.
6. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46(3):795-806.
7. Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355-61.
8. Baddini-Martinez J, Pereira CA. How many patients with idiopathic pulmonary fibrosis are there in Brazil? J Bras Pneumol. 2015;41(6):560-1.
9. Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, et al. Blue journal conference. Aging and susceptibility to lung disease. Am J Respir Crit Care Med. 2015;191(3):261-9.
10. Spagnolo P, Rossi G, Cavazza A. Pathogenesis of idiopathic pulmonary fibrosis and its clinical implications. Expert Rev Clin Immunol. 2014;10(8):1005-17.
11. Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med. 2014;189(10):1161-72.
12. Ley B, Collard HR, King TE. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(4):431-40.
13. Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am J Respir Crit Care Med. 2016;194(3):265-75. 14. Brown AW, Fischer CP, Shlobin OA, Buhr RG, Ahmad S, Weir NA, et al. Outcomes after hospitalization in idiopathic pulmonary fibrosis: a cohort study. Chest. 2015;147(1):173-9. 15. Nathan SD, Meyer KC. IPF clinical trial design and endpoints. Curr Opin Pulm Med. 2014.
16. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184(12):1382-9.
17. Lama VN, Flaherty KR, Toews GB, Colby TV, Travis WD, Long Q, et al. Prognostic value of desaturation during a 6-minute walk test in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003;168(9):1084-90.
18. Stephan S, de Castro Pereira CA, Coletta EM, Ferreira RG, Otta JS, Nery LE. Oxygen desaturation during a 4-minute step test: predicting survival in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2007;24(1):70-6.
19. Mogulkoc N, Brutsche MH, Bishop PW, Greaves SM, Horrocks AW, Egan JJ, et al. Pulmonary function in idiopathic pulmonary fibrosis and referral for lung transplantation. Am J Respir Crit Care Med. 2001;164(1):103-8.
20. Collard HR, King TE, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168(5):538-42.
21. Flaherty KR, Thwaite EL, Kazerooni EA, Gross BH, Toews GB, Colby TV, et al. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax. 2003;58(2):143-8.
22. Russell AM, Adamali H, Molyneaux PL, Lukey PT, Marshall RP, Renzoni EA, et al. Daily Home Spirometry: An Effective Tool for Detecting Progression in Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2016;194(8):989-97.
23. Fukuda CY, Soares MR, Pereira CAC. A Score Without DLCO for Estimating Survival in Idiopathic Pulmonary Fibrosis. Am J Resp Crit Care Med. 2018;197:A2535.
24. Dal Corso S, Duarte SR, Neder JA, Malaguti C, de Fuccio MB, de Castro Pereira CA, et al. A step test to assess exercise-related oxygen desaturation in interstitial lung disease. Eur Respir J. 2007;29(2):330-6.
25. Mancuzo EV, Soares MR, Pereira CAC. Six-minute walk distance and survival time in patients with idiopathic pulmonary fibrosis in Brazil. J Bras Pneumol. 2018;44(4):267-72.
26. Costa CM, Neder JA, Verrastro CG, Paula-Ribeiro M, Ramos R, Ferreira EM, et al. Uncovering the mechanisms of exertional dyspnoea in combined pulmonary fibrosis and emphysema. Eur Respir J. 2020;55(1).
27. Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156(10):684-91.
28. Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, et al. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med. 2003;167(7):962-9.
29. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Ascertainment of individual risk of mortality for patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(4):459-66.
30. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-82.
31. Troy LK, Grainge C, Corte TJ, Williamson JP, Vallely MP, Cooper WA, et al. Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir Med. 2020;8(2):171-81.
32. van Manen MJ, Geelhoed JJ, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2017;11(3):157-69.
33. Kreuter M, Swigris J, Pittrow D, Geier S, Klotsche J, Prasse A, et al. The clinical course of idiopathic pulmonary fibrosis and its association to quality of life over time: longitudinal data from the INSIGHTS-IPF registry. Respir Res. 2019;20(1):59.
34. Hilberg O, Bendstrup E, Ibsen R, Løkke A, Hyldgaard C. Economic consequences of idiopathic pulmonary fibrosis in Denmark. ERJ Open Res. 2018;4(2).
35. Loveman E, Jones J, Clegg AJ, Picot J, Colquitt JL, Mendes D, et al. The clinical effectiveness and cost-effectiveness of ablative therapies in the management of liver metastases: systematic review and economic evaluation. Health Technol Assess. 2014;18(7):vii-viii, 1-283.
36. King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083-92.
37. Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015;192(2):e3-19.
38. Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug Treatment of Idiopathic Pulmonary Fibrosis: Systematic Review and Network Meta-Analysis. Chest. 2016;149(3):756-66.
39. van Manen MJG, Birring SS, Vancheri C, Vindigni V, Renzoni E, Russell AM, et al. Effect of pirfenidone on cough in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2017;50(4).
40. Maher TM, Strek ME. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir Res. 2019;20(1):205.
41. Baddini-Martinez J, Baldi BG, Costa CH, Jezler S, Lima MS, Rufino R. Update on diagnosis and treatment of idiopathic pulmonary fibrosis. J Bras Pneumol. 2015;41(5):454-66.
Sorry, the comment form is closed at this time.